
Sequencing after chromatin immunoprecipitation (ChIP seq) is a genome-wide analysis technique for DNA binding proteins, histone modifications, or nucleosomes. Due to the significant advancement of second-generation sequencing technology, ChIP-seq has higher resolution, lower noise, and more extensive coverage than its original version, ChIP-chip. As a result, ChIP-seq has become an indispensable tool for studying gene regulation and epigenetic mechanisms to reduce sequencing costs.
1. Principle
Cells are treated with formaldehyde to cross-link the target protein with DNA, and the cross-linked chromatin is broken into small fragments by ultrasound, generally in the range of 200-600bp. Then, the specific recognition reaction of antigen and antibody is used to precipitate the DNA fragments that bind to the target protein. Finally, cross-linking and PCR amplification of the purified DNA, high-throughput sequencing, and final comparison with the existing genome sequence determine the sequence of DNA and protein binding.
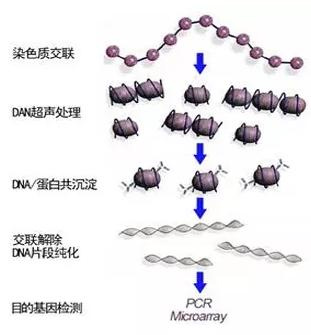
2. Problem analysis
2.1. The influence of formaldehyde cross-linking on subsequent analysis of results?
Since Orlando V and others first invented ChIP technology, the core steps have not changed much after more than ten years. However, ChIP-seq technology has certain shortcomings, such as formaldehyde cross-linking. Although formaldehyde is a highly absorbent cross-linking agent, its reactivity is limited to amines. Its cross-linking efficiency is low; for mammalian cells, its maximum cross-linking efficiency is only 1%. Therefore, the initial amount required for formaldehyde cross-linking of cells is enormous, and thus the technique is challenging to apply to micro-cell and single-cell samples. The article In vivo formaldehyde cross-linking: it is time for black-box analysis published by Oxford University Press pointed out that certain proteins such as repressor protein and NF-κB cannot be cross-linked to DNA by formaldehyde. Studies have shown that Proteins with a residence time shorter than 5 seconds cannot be cross-linked with formaldehyde on DNA. Secondly, formaldehyde will cause many other irrelevant proteins to cross-link to DNA, affecting subsequent analysis data. It has been reported that formaldehyde cross-linking can trigger the DNA damage response mechanism, thereby changing the chromosome composition and then biasing the results of ChIP. In addition, since the cross-linking reaction is reversed under heating and low pH, the stability of the cross-linked complex between DNA protein and white matter is also a concern. Therefore, the extent to which formaldehyde can reflect the distribution of proteins in cells is still uncertain.
According to the presence or absence of formaldehyde cross-linking steps, CHIP technology can be divided into two types. One is ChIP with formaldehyde cross-linking, namely X-ChIP (cross-linking and mechanical shearing ChIP); the other is without cross-linking. ChIP, or N-ChIP (native-ChIP); Compared with X-ChIP, N-ChIP technology has a series of advantages, including high resolution (MNase makes the fragmentation of chromosomes as small as nucleosomes). It avoids the enrichment of non-specific proteins on DNA caused by formaldehyde cross-linking, avoids the hiding of antigen epitopes by formaldehyde cross-linking, and reduces steps that reduce the loss of samples. However, due to the use of MNase, N-ChIP is only suitable for the study of histone modifications, and in most cases, cannot be used for the analysis of transcription factors.
2.2. Comparison of ultrasonic interruption and enzyme cleavage methods?
Enzymes: The most commonly used enzymes such as MNase, namely: micrococcal nuclease, is a nuclease that can degrade the DNA sequence of the nucleosome junction region, originally isolated from Staphylococcus aureus. MNase digests chromatin to release individual nucleosomes.
The MNase enzymatic hydrolysis method has certain limitations. First, MNase is biased towards cutting A/T base sites, resulting in lower nucleosome expression in the A/T enriched region; secondly, MNase cannot be in the nucleosome. The body boundary is precisely cut, which leads to a difference in determining the open position of chromatin and the actual situation; moreover, MNase is biased towards digesting fragile nucleosomes. Experimental evidence in different species shows that fragile nucleosomes occupy gene promoters and transcription termination sites. In contrast, fragile nucleosomes can only be detected when the MNase concentration is low. Furthermore, the digestion time is short, so it is challenging to Quantify fragile nucleosomes to the relative abundance of stable nucleosomes.
Ultrasound interruption is not as gentle as the enzymatic lysis method. Due to the unevenness of the interruption, the background noise of the sequencing results is high, which affects the subsequent data analysis. Due to the length of the article, I won’t repeat it here.
So the choice of enzymatic hydrolysis or ultrasound interruption depends on the situation. You can refer to the following suggestions:
If the protein under study is expressed in high abundance and binds to DNA as tightly as histones, then the sample does not need to be cross-linked, and enzymatic hydrolysis can be used.
Suppose the protein under study is expressed in low abundance or does not bind to DNA tightly, such as transcription factors. In that case, it is often necessary to fix the sample with cross-linking reagents to stabilize the morphology of the protein and DNA. In this case, it is best to use ultrasound for fragmentation.
3. Expand
3.1 ChIP technology and its principle suitable for micro-cell level
(1) CUT-Tag technology
CUT-Tag can be used to study the binding sites of transcription factors and the openness of DNA simultaneously. It can complete all steps from cell to library building within one day, and it has the characteristics of high resolution and low noise. In addition, the initial cell dosage can be as low as 50.
principle
Via antigen-antibody specific reaction, adding a particular antibody to bind to the target protein on the chromosome, adding a second antibody to attach to the antibody to recruit more pA-Tn5 transposase complexes to the binding site of the target protein to the DNA sequence, the transposable enzyme complex cleaves the open area of the chromatin and joins junctions at both ends of the DNA fragment. Thus, the library is constructed and can be directly sequenced for comparison with existing genomic sequences, the binding site of the target protein to the DNA is known.
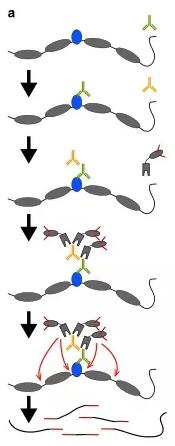 (2) ChIL-seq
(2) ChIL-seq
ChIL (chromatin integration labeling), the chromatin integration labeling technology, can be used to study the binding sites of transcription factors and the openness of DNA simultaneously. The initial cell dosage is 100-1000 cells. ChIL-seq avoids the low recovery rate caused by antibody precipitation in traditional ChIP-seq technology and is especially suitable for adherent cells. For active histone markers such as H3K4me3 and H3K27ac, the starting cell dosage can be reduced to the single-cell level.
principle
Add cells to a 96-well plate, fix, stain, and add primary antibody to bind to the target protein, then add ChIL probe (consisting of the secondary antibody and ChIL DNA). The ChIL DNA in the probe is integrated into the target by Tn5 transposase Near the genomic DNA where the protein is located, then T7 RNA polymerase initiates transcription via the promoter in ChIL DNA. Uses the genomic DNA as a template to synthesize RNA, digests, and cleaves with DNase I to release RNA, and builds a library with purified RNA for sequencing.
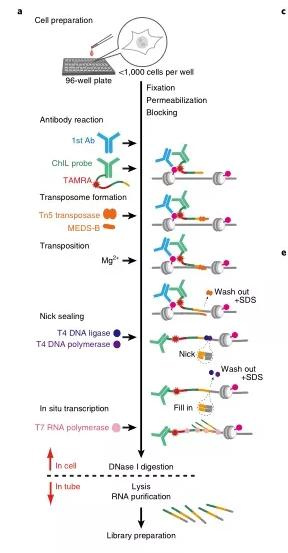
(3) Drop-ChIP
Drop-ChIP: Via a specific microfluidic device, the resolution can reach the single-cell level. This technology can study transcription factor binding sites and histone modifications at the single-cell level and learn the degree of chromatin variation between different cells from a cell-specific perspective. We believe that integrating single-cell chromatin and single-cell expression data can enable a more precise coupling of regulatory elements and target genes and a deeper understanding of their functional dynamics and relationships.
principle
First, mix the cells to be studied with the lysate and MNase for chromatin digestion. In addition, a droplet containing many different joints is designed and reacted on the microfluidic device so that each cell droplet is connected to a joint droplet. Next, mix and mix with the buffer droplet containing DNA ligase at the same time. In this process, the linker sequence is automatically connected to both ends of the lysed chromatin fragments, and the cells are then lysed. Specific antibodies are used to precipitate the target protein, and the enriched DNA is sequenced. Align with the existing genome sequence to know the target protein action site.
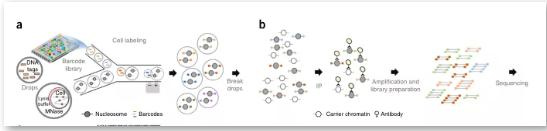
references:
[1]. 1: Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, Henikoff S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019 Apr 29;10(1):1930.
[2]. Harada A, Maehara K, Handa T, Arimura Y, Nogami J, Hayashi-Takanaka Y,
Shirahige K, Kurumizaka H, Kimura H, Ohkawa Y. A chromatin integration labeling method enables epigenomic profiling with lower input. Nat Cell Biol. 2019 Feb;21(2):287-296.
[3]. Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, Bernstein BE.
Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol. 2015 Nov;33(11):1165-72
Disclaimer: Article source-YiGene (WeChat public account) Author: e-gene, to spread knowledge, beneficial learning, and research. If the content is wrong, please criticize and correct it. The reprint is only for reference, study, and transfer of useful information. The copyright belongs to the original author. If you infringe on rights, please contact to delete.