Abstract
Tissues are complex mixtures of different cell subtypes, and this diversity is increasingly characterized using high-throughput single cell analysis methods. However, these efforts are hindered, as tissues must first be dissociated into single cell suspensions using methods that are often inefficient, labor-intensive, highly variable, and potentially biased towards certain cell subtypes. Here, we present a microfluidic platform consisting of three tissue processing technologies that combine tissue digestion, disaggregation, and filtration. The platform is evaluated using a diverse array of tissues. For kidney and mammary tumor, microfluidic processing produces 2.5-fold more single cells. Single cell RNA sequencing further reveals that endothelial cells, fibroblasts, and basal epithelium are enriched without affecting stress response. For liver and heart, processing time is dramatically reduced. We also demonstrate that recovery of cells from the system at periodic intervals during processing increases hepatocyte and cardiomyocyte numbers, as well as increases reproducibility from batch-to-batch for all tissues.
Introduction
Tissues are highly complex ecosystems containing a diverse array of cell subtypes. Significant variation can also arise within a given subtype due to differences in activation state, genetic mutations, epigenetic distinctions, stochastic events, and microenvironmental factors1,2. This has led to a rapid growth in studies attempting to capture cellular heterogeneity, and thereby gain a better understanding of tissue and organ development, normal function, and disease pathogenesis3,4,5,6,7,8,9. For example, in the context of cancer, intratumor heterogeneity is a key indicator of disease progression, metastasis, and the development of drug resistance10,11,12,13,14. High-throughput single-cell analysis methods such as flow cytometry, mass cytometry, and single-cell RNA sequencing (scRNA-seq) are ideal for identifying single cells in a comprehensive manner based on molecular information15,16, and these methods have already begun to transform our understanding of complex tissues by enabling identification of previously unknown cell types and states8,17,18,19. However, a critical barrier to these efforts is the need to first process tissues into a suspension of single cells. Current methods involve mincing, digestion, disaggregation, and filtering that are labor-intensive, time consuming, inefficient, and highly variable20,21. Thus, new approaches and technologies are critically needed to ensure reliability and wide-spread adoption of single-cell analysis methods for tissues. This would be particularly important for translating single-cell diagnostics to human specimens in clinical settings. Moreover, improved tissue dissociation would make it faster and easier to extract primary cells for ex vivo drug screening, engineered tissue constructs, and stem/progenitor cell therapies22,23,24,25. Patient-derived organ-on-a-chip models, which seek to recapitulate complex native tissues for personalized drug testing, are a particularly exciting future direction that could be enabled by improved tissue dissociation22,26,27,28,29,30.
scRNA-seq has recently emerged as a powerful and widely adaptable analysis technique that provides the full transcriptome of individual cells. This has enabled comprehensive cell reference maps, or atlases, to be generated for normal and diseased tissues, as well as identification of previously unknown cell subtypes or functional states31,32. For example, an atlas recently generated for normal murine kidney uncovered a new collecting duct (CD) cell with a transitional phenotype and an unexpected level of cellular plasticity4. Moreover, an atlas of primary human breast epithelium linked distinct epithelial cell populations to known breast cancer subtypes, suggesting that these subtypes may develop from different cells of origin3. For melanoma, scRNA-seq was used to identify three transcriptionally distinct states, one of which was drug sensitive, and further demonstrated that drug resistance could be delayed using computationally optimized therapy schedules33. While scRNA-seq is clearly a powerful diagnostic modality, the process of breaking down the tissue into single cells can introduce confounding factors that may negatively influence data quality and reliability. One factor is the lack of standardization, which can lead to substantial variation across different research groups and tissue types. Another significant concern is that incomplete breakdown could bias results toward cell types that are easier to liberate. A recent study utilizing single-nuclei RNA sequencing with murine kidney samples found that endothelial cells and mesangial cells (MC) were underrepresented in scRNA-seq data34. Finally, lengthy enzymatic digestion times have been shown to alter transcriptomic signatures and generate stress responses that interfere with cell classification35,36,37,38,39. Addressing these concerns would help propel the exciting field of scRNA-seq into the future for tissue atlasing and disease diagnostics.
Microfluidic technologies have advanced the fields of biology and medicine by miniaturizing devices to the scale of cellular samples and enabling precise sample manipulation40,41,42,43,44. Most of this work has focused on manipulating and analyzing single cells44,45,46,47,48. Only a small number of studies have addressed tissue processing, and even fewer have focused on breaking down tissue into smaller constituents49,50,51. We previously developed a microfluidic device that specifically focused on breaking down cellular aggregates into single cells52,53. This dissociation device contained a network of branching channels that progressively decreased in size down to ~100 µm, and contained repeated expansions and constrictions to break down aggregates using shear forces. We then developed a device for on-chip tissue digestion using the combination of shear forces and proteolytic enzymes54. Finally, we developed a filter device containing nylon mesh membranes that removed large tissue fragments, while also dissociating smaller cell aggregates and clusters55. The microfluidic digestion, dissociation, and filter devices each enhanced single cell recovery when operated independently. To date, however, we have not combined these technologies to maximize performance and execute a complete tissue processing workflow on-chip. Moreover, we have not validated microfluidically processed cell suspensions using scRNA-seq.
In this work, we present a microfluidic platform comprised of three different tissue-processing technologies that enhances the breakdown of tissue and produces single-cell suspensions that are immediately ready for downstream single-cell analysis or other use. First, we design a digestion device that can be loaded with minced tissue and operated with minimal user interaction. Next, we integrate the dissociation and filter technologies into a single unit, and optimize the two-device platform using murine kidney to produce single cells more quickly and in higher numbers than traditional methods. Using the optimized protocol, we evaluate different tissue types using two single-cell analysis methods. For murine kidney and breast tumor tissues, microfluidic processing can produce >2-fold more epithelial cells and leukocytes, and >5-fold more endothelial cells. Using scRNA-seq, we show that device processed samples are highly enriched for endothelial cells, fibroblasts, and basal epithelium. We also demonstrate that stress responses are not induced in any cell type, and can even be reduced if shorter processing times are employed. For murine liver and heart, significant single cell numbers are obtained after only 15 min, and even as short as 1 min. Interestingly, we find that substantially more hepatocytes and cardiomyocytes are obtained if sample is recovered at discrete intervals, most likely because these cell types are sensitive to shear forces. Importantly, the microfluidic platform can significantly shorten processing time or enhance single cell recovery for all tissue types studied, and in some cases accomplish both, while increasing batch-to-batch reproducibility and maintaining viability. Furthermore, the entire tissue processing workflow is performed in an automated fashion. Thus, our microfluidic platform holds exciting potential to advance diverse applications that require the liberation of single cells from tissues.
Results
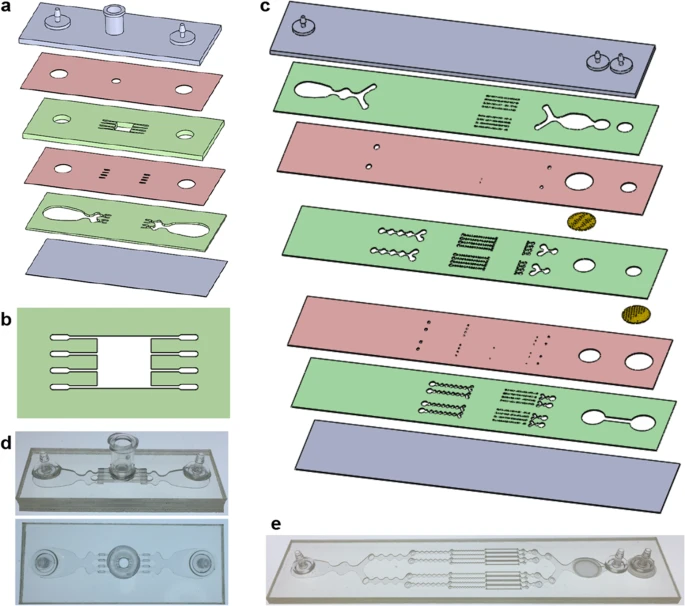
We designed a digestion device that would not require manual device assembly. Instead, minced tissue is loaded through a port at the top of the device, which can then be sealed using a cap or stopcock. Scalpel mincing of tissue into ~1 mm3 pieces is ubiquitous, and therefore this format will be compatible with a wide array of tissue types and dissociation protocols. The full design layout of the minced tissue digestion device is shown in Fig. 1a, including the loading port, a chamber that retains the tissue in place, and fluidic channels that administer fluid shear forces and deliver proteolytic enzymes. These features were arranged across six layers of hard plastic, including two fluidic channel layers, two “via” layers, a top end cap with hose barbs and loading port, and a bottom end cap. The tissue chamber is in the uppermost fluidic layer, directly beneath the loading port and a 2.5 mm diameter via, and a detailed schematic is shown in Fig. 1b. We employed a square geometry, with 5 mm length and width, to allow tissue to be evenly distributed during loading. Chamber height was 1.5 mm, slightly larger than minced tissue, to prevent clogging during sample loading and device operation. Fluidic channels were placed upstream and downstream of the tissue chamber, and in both cases, we employed four channels that were 250 µm wide. The symmetric channel design was chosen for the minced format because there is a greater emphasis on prevention of clogging. We also extended channel length to 4 mm to prevent larger tissue pieces from squeezing all the way through the channels, but flared the end to make it easier to connect with the underlying via layer.